Introduction
This form of anemia is due to a genetic abnormality in the synthesis of the hemoglobin molecule of the red blood cell. In the adult the sickle cell hemoglobin with the name hemoglobin S replaces the normal hemoglobin A. The difference is an exchange of the amino acid valine by glutamic acid from the genetic abnormality. This genetic abnormality occurs almost exclusively in blacks. About 0.3% of black people in the US have this dominant form of genetic constellation. People with the pure (homozygous) form of sickle cell anemia are symptomatic.
Heterozygous sickle cell anemia
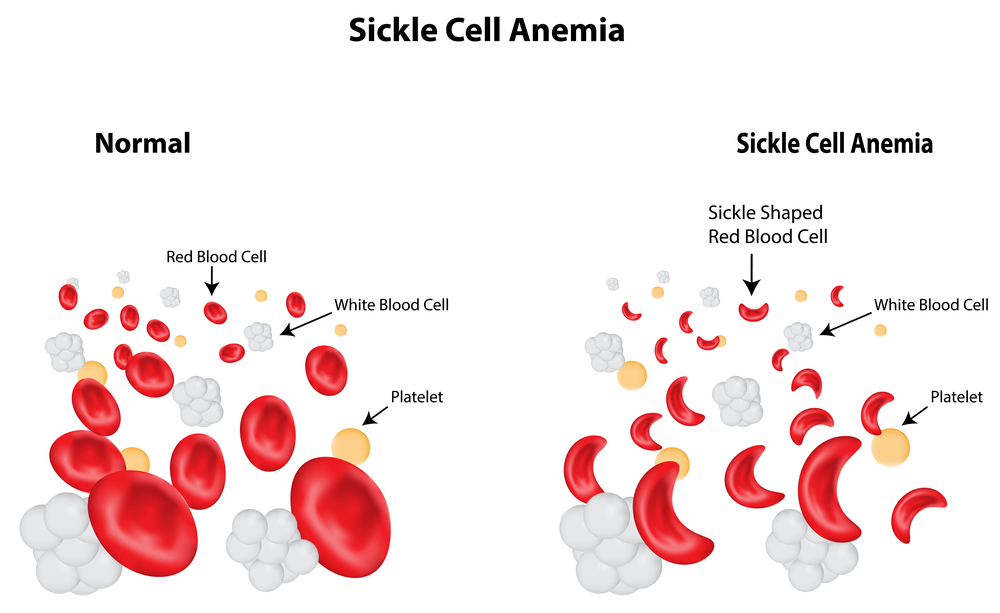
About 10% of blacks in the US have a milder version (heterozygous for the sickle cell gene) and they are not anemic. Patients with this trait need to be under the care of a hematologist who can help to alleviate some of the symptoms. The red blood cells are distorted into sickle shape forms (seen through the microscope). These are more rigid than normal red blood cells and plug capillaries leading to infarction of tissues.
Symptoms of sickle cell anemia
It is mostly the pure form (homozygous form) of sickle cell anemia, which causes symptoms. The skin is pale and signs of jaundice may be present. A large heart is common (cardiomegaly) and the doctor can hear a murmur with the stethoscope (“ejection murmur”). There can be acute pain (“crises”) in the bones, the spleen, the lungs and the kidneys. On a microscopic level the pathologist sees infarctions in the tissues that result in ischemia (lack of oxygen). This is the reason for the painful bouts. However, it is not always clear why this acute pain occurs. Some of the precipitating factors can be an infection, fever or a local trauma. But many times the doctor can not find an apparent reason. The doctor will often find an enlarged spleen and liver (called “hepatosplenomegaly”). As this is a genetic condition, it occurs already in infants and toddlers.
Autosomal form of sickle cell anemia
In the more severe autosomal forms, the growth pattern will be influenced leading to a characteristic appearance with a short trunk, long legs and arms and a skull shaped like a tower. Chronic leg ulcers that have a punched-out appearance are commonly found in the area of the ankles. By the time the patients are adults they have experienced so many bouts of mini-infarctions in liver and spleen that the organs have become fibrotic and the spleen that is particularly vulnerable will be small. Patients with sickle cell anemia are more prone to develop gall stones (cholelithiasis). Avascular necrosis of the femoral head is another common complication.
Acute chest syndrome in children
children an acute chest syndrome is particularly common, which leads to complications with death in 10%. When this occurs, it usually starts with a fever, chest pains and infiltrates of the lungs visible on X-rays. These infiltrates start in the base of the lungs (bilateral lower lobe infiltrates) and there may be water in the chest cavities (pleural effusions). Bacterial pneumonia may set in as well. As these changes are from mini-infarcts in the tissues of the lungs and the heart, a severe lack of oxygen (hypoxemia) develops rapidly. In chronic recurrent bouts patients may develop pulmonary hypertension as another complication. In young men priapism can develop, which may lead to erectile dysfunction. Also, ischemic strokes and vasculitis in the brain may occur.
Diagnostic tests
There is a rapid screening test (tube test), which relies on the fact that hemoglobin S is less soluble than normal hemoglobin A. The usual screening tests for hemolytic anemias are done, which will show a normocytic anemia with reticulocytosis (more than 10%). Hemoglobin electrophoresis is done, which will show the hemoglobin S peak. A blood smear will show the sickle cell, which is a sickle shaped red blood cell. The hemolysis leads to an elevate bilirubin level, which can be measured in a blood sample. The urine will test positive for urobilinogen, which is a breakdown product of bilirubin. The patient would by now referred to a hematologist (or if a child to a pediatrician). Further tests may be required such as a bone marrow.
Bone marrow can become aplastic
After a painful crisis with infection the bone marrow may have become aplastic meaning that precursor cells of blood cells are missing. Hematuria (blood in urine) can occur during a painful crisis, particularly when kidney pains are present. This can be detected with a dipstick. Electrophoresis is useful in detecting the difference between the more severe homozygous state of sickle cell anemia and the less problematic heterozygous state. The homozygous state shows only hemoglobin S on electrophoresis, with varying amounts of hemoglobin F (which is also abnormal in toddlers, older children or adults). In contrast with electrophoresis the heterozygous state shows more hemoglobin A than hemoglobin S. In the meantime, geneticists have developed more direct PCR techniques that show the genetic defect directly.
Sickle Cell Anemia
Treatment and Prognosis
Patients with the homozygous state of sickle cell anemia need close medical supervision. They can expect to live longer than age 50 (which was rare in the past). With the better understanding of the cellular mechanisms (pathophysiology) of this anemia the physician can now modify the severity of the crises with less tissue damage. The common complications in homozygous patients that can cause death occur in association with the acute chest syndrome, pulmonary emboli, and infarctions of the heart, liver, bone marrow, lungs or brain and secondary infections. Unfortunately, there is no anti-sickling drug available. As a result, the physician will mainly treat supportively during the approximate 5 days of crisis where pain and fever come to a peak. Strong pain relievers are given. Fluid intake and output are monitored and balanced. The patient is monitored for oxygenation of the blood.
Complications with sickle cell anemia
Removal of the spleen has not made a difference in survival, but sometimes has to be done, if complications (ruptured spleen) set in. Hospitalization is required for serious infections that require antibiotics, for acute chest syndrome, or if the anemia is so severe (hemoglobin count less than 5 grams/deciliter) that transfusions are required. Transfusions need to be spaced as much apart as possible, but close enough to avoid clots in the brain (recurrent cerebral thrombosis) in children less than 18 years where this is more common. Other reasons to hospitalize are aplastic crises, acute stroke, acute chest syndrome or specific organ failures. When oxygen tension is less than 65mm mercury (measured by blood gases) the physician likely will also hospitalize the patient. Because of the recurrent immune weakness, the patient is vaccinated against the major bacterial strains: H. influenzae vaccine, meningococcal vaccine and pneumococcal vaccine.
Various treatment steps that are useful
Prophylactic antibiotics (oral penicillin from the age of 4 months to 6 years) have also contributed to less childhood mortality. Folate is given daily (1 milligram orally). Hydroxyurea works by increasing fetal hemoglobin (hemoglobin F), which cuts the painful crises by 50%. It also helps to reduce the frequency of the need for transfusions and reduces the severity of the acute chest syndrome. The amount of hydroxyurea is dependent on the hemoglobin F level, which can be monitored with blood tests. The beneficial effect may be amplified when this is combined with erythropoietin (about 50,000 units per week). Fortunately, with good medical care patients with heterozygous trait sickle cell anemia (10% of blacks in the US) have much less complications and a normal life expectancy.
Cure of sickle cell anemia
The newest line of research is working on a cure for sickle cell anemia and some people have already been cured. Just a few years back this type of research would never have been thought possible, but now it is becoming a reality.
References
1. Merck Manual (Home edition): Anemia
2. Noble: Textbook of Primary Care Medicine, 3rd ed., Mosby Inc. 2001
3. Goldman: Cecil Medicine, 23rd ed., Saunders 2007: Chapter 162 – APPROACH TO THE ANEMIAS