Introduction
First, hemoglobinopathies are genetic abnormalities of the hemoglobin molecule, which is a complicated protein that provides structural integrity to the red blood cells and allows the red blood cells to transport oxygen to the tissues and carbon dioxide from the tissues to the lungs in an optimal way. Second, each hemoglobin molecule has 4 iron attachment sites for oxygen molecules as depicted in the illustration below. Third, from this it is easy to see that abnormalities of the hemoglobin molecule will create problems with regard to oxygen transport from the lungs into the tissues.
Inherited hemoglobin defects
There are many inherited defects of the hemoglobin molecule and this translates into such abnormalities as sickle cell anemia, the hemoglobin C disease, hemoglobin E disease, hemoglobin S-C disease and thalassemia. See the links attached to this page for more details on these conditions. As a brief overview it is important to note that during fetal life there is a different hemoglobin present (hemoglobin F), which is better suited for the fetal blood circulation where the lung function is taken care of by the placenta. In the adult less than 2% of the total hemoglobin consists of hemoglobin F, the remainder is hemoglobin A, which is the normal hemoglobin. In general, with hemoglobinopathies usually a higher percentage of hemoglobin F is present in children and adults, which is a clue during the diagnostic work-up.
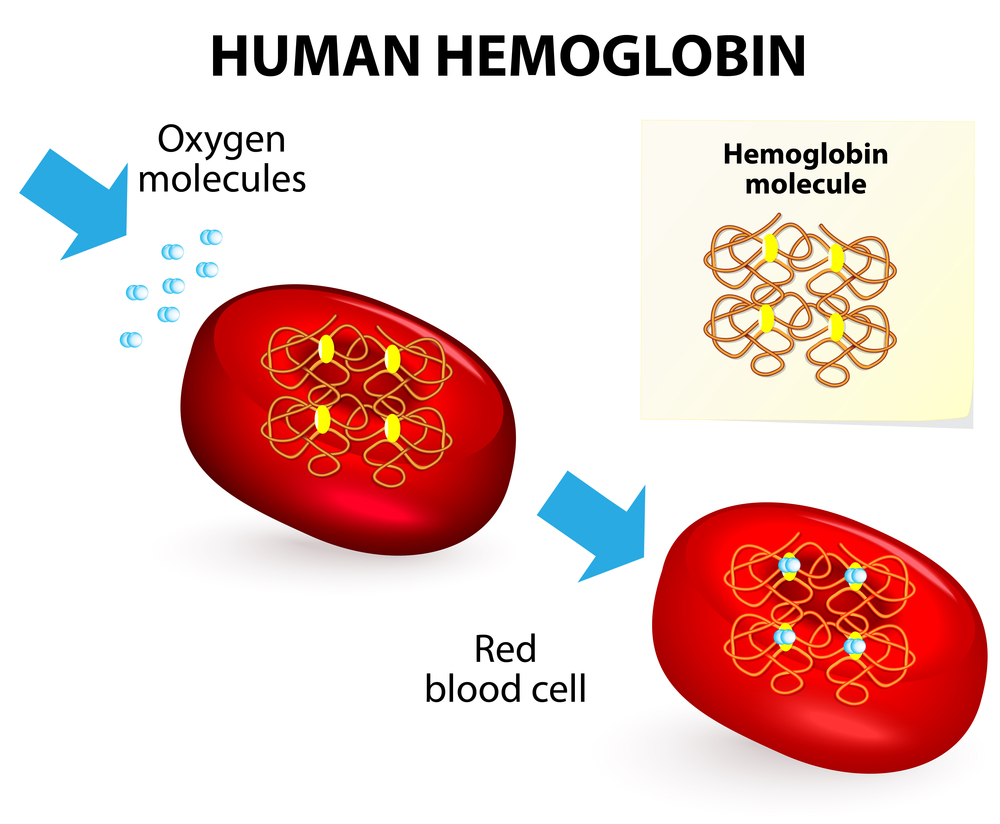
Hemoglobinopathies (Oxygen Transport)
Hemoglobin C Disease
To clarify, about 2.5% of blacks in the US have this trait. It is a hemoglobinopathy where abnormal hemoglobin is produced leading to an anemia similar to sickle cell anemia, but not as severe. Unlike sickle cell disease hemoglobin C disease (in homozygotes) does not produce the abdominal crises typical for sickle cell disease. On the other hand, an enlarged spleen (splenomegaly) is typically present. The laboratory findings show a moderate anemia with 35 to 100% target cells. The smear also shows spherocytes and red blood cells that contain crystals. The red blood cells do not show sickling, but some may be nucleated. The decisive test is by electrophoresis that shows the hemoglobin C. Heterozygotes are symptom free, but their red blood cells show central targeting. Even the more symptomatic homozygotes usually do not have severe enough symptoms to warrant treatment.
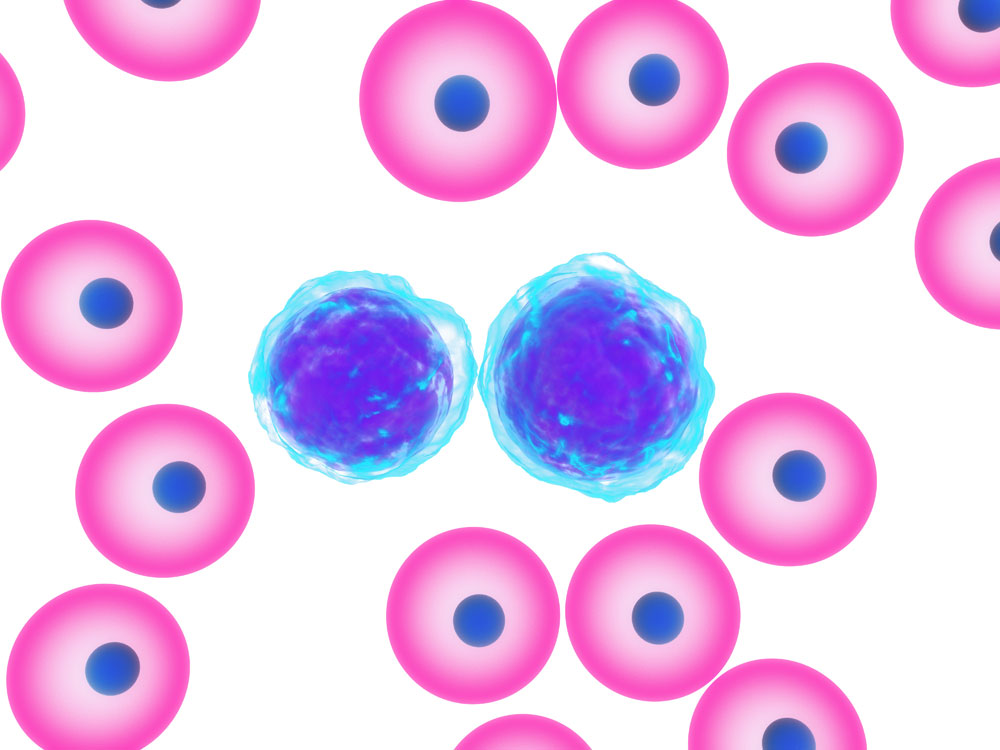
Hemoglobinopathy With Target Cells
Hemoglobin S-C Disease
This hemoglobinopathy can occur in a child where one parent has sickle cell anemia (hemoglobin S) and the other parent has hemoglobin C disease. It is most commonly found in blacks. The symptoms are similar to a patient with sickle cell anemia, but much milder. In brief, frank hematuria (blood in urine), spontaneous hip bone fractures (aseptic necrosis of the femoral head) and retinal bleeding (retinal hemorrhages) are common. On the whole, the blood tests show target cells in blood smears with the occasional sickling as well, and hemoglobin S-C on electrophoresis.
Hemoglobin E Disease
This hemoglobinopathy is quite common among blacks and Southeast Asians (more than 15% carry the homozygous form), but is rare among the Chinese. Depending on what genetic combination the patient has the clinical presentation will be different. The mildest form is the heterozygote with hemoglobin AE who will be asymptomatic. On the other hand, a homozygote with hemoglobin E disease will have a mild anemia with target cells. The most severe clinical condition is a patient heterozygous for hemoglobin E and beta-thalassemia. These patients have some symptoms of thalassemia and have an enlarged spleen. These patients require a close follow-up by a hematologist. Electrophoresis is a tool that identifies all of these various genetic forms of hemoglobin E disease.
Hemoglobin S-Beta-Thalassemia Disease
This is a hemoglobinopathy that has both the genetic defect of sickle cell anemia and of beta-thalassemia. It occurs when children are born to parents who carry these two different genes. As the diseases run in overlapping population groups, this combination is not infrequent. Microcytic anemia is mild to moderate. There will be some sickled red blood cells on a blood smear. Quantitative hemoglobin analysis shows that hemoglobin A2 is greater than 3%, hemoglobin F is increased, hemoglobin A decreased or even absent. Electrophoresis shows the exact hemoglobin constellation. Milder cases often do not need to be treated; others who are symptomatic are treated as explained under sickle cell anemia.
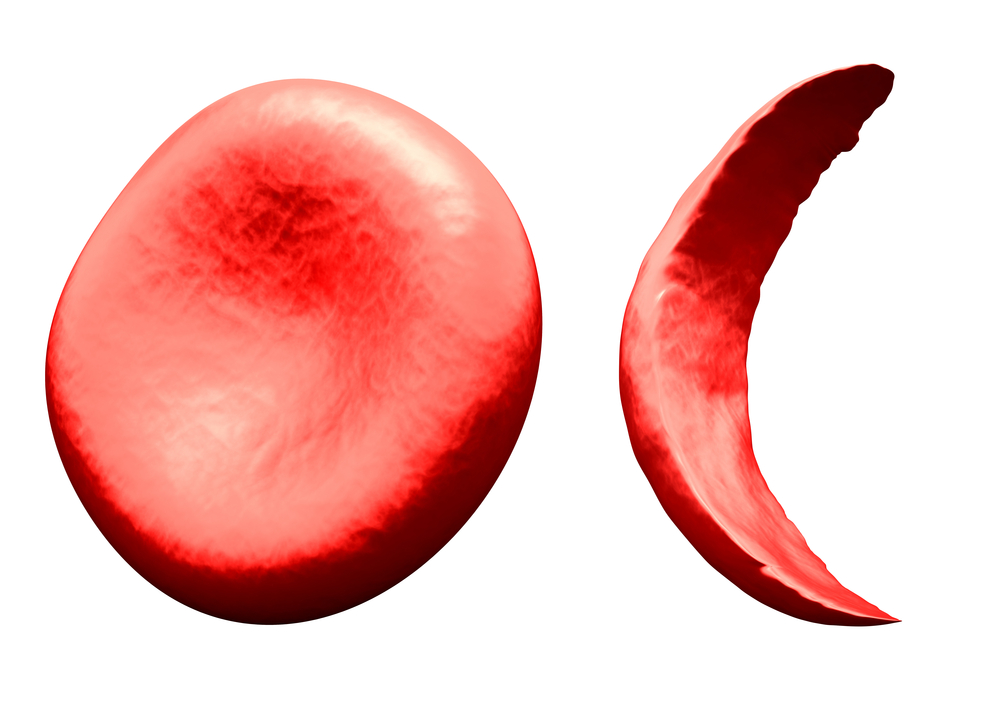
Hemoglobinopathies (Normal Red Blood Cell Left, Sickle Cell Right)
References
1. Merck Manual (Home edition): Anemia
2. Noble: Textbook of Primary Care Medicine, 3rd ed., Mosby Inc. 2001
3. Goldman: Cecil Medicine, 23rd ed., Saunders 2007: Chapter 162 – APPROACH TO THE ANEMIAS